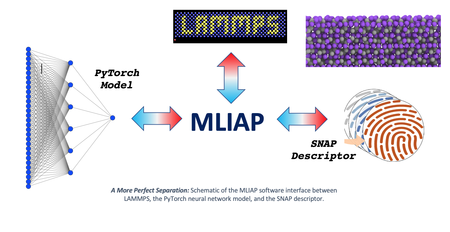
Researchers at Sandia and Los Alamos National Laboratories have discovered a new way to implement machine learning (ML) interatomic potentials in the LAMMPS molecular dynamics code. This makes it much easier to prototype and deploy ML models in LAMMPS and provides access to a vast reservoir of existing ML libraries. The key is to define an interface (MLIAP) that separates the calculation of atomic fingerprints from the prediction of energy. The interface also separates the atomic descriptors and energy models from the specialized LAMMPS data structures needed for efficient simulations on massively parallel computers. Most recently, a new model class has been added to MLIAP that provides access to any Python-based library, including the PyTorch neural network framework. This advancement was made under the DOE SciDAC/FES FusMatML project for the application of machine learning to atomistic models for plasma-facing materials in fusion reactors.
More information on the LAMMPS package for machine-learned interatomic potentials can be found here: LAMMPS MLIAP Package