Luke Shulenburger (1641) and Andrew Baczewski (1425)
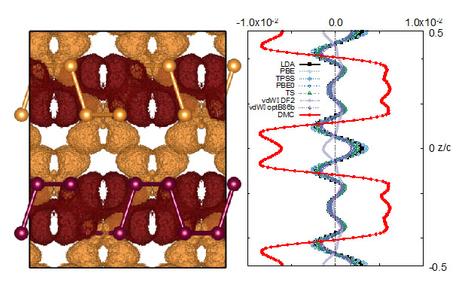
Recently, we published work in Nano Letters (Impact Factor 13.592) detailing the results of quantum Monte Carlo (QMC) calculations from our BES-funded investigation of the properties of phosphorene, a single-layer form of black phosphorus [1]. Black phosphorus is a layered form of phosphorus in which highly anisotropic 2D sheets are held together by strong covalent bonds, and stacks of these sheets are nominally held together by the van der Waals’ interaction [2]. A single layer of black phosphorus can be exfoliated yielding phosphorene, a material experiencing intense research for applications in electronics due to its direct band gap, high carrier mobility, and strain-sensitivity [3]. The relationship between phosphorus and phosphorene, is analogous to that of graphite and graphene, however, graphene has no band gap, which has impeded some electronics applications. Our QMC calculations indicate that we can accurately compute experimentally measured structural parameters of the bulk system [2], giving us confidence in our predictions of the as-yet-unmeasured single- and few-layer structure and the exfoliation energy required to mechanically remove a single sheet of phosphorene. We also predict that stacking fault defects will be energetically unfavorable in few-layer preparations of phosphorene. As few-layer phosphorene is also of interest for applications, this is critical enabling information for experiment.
One unexpected outcome of our study was uncovering a previously unknown inaccuracy in Density functional theory (DFT). DFT is, by far, the most popular ab initio electronic structure method for materials science. However, while our QMC calculations predict significant charge reorganization in going from single- to few-layer phosphorene, or single-layer phosphorene to bulk black phosphorus, DFT completely fails to predict any charge reorganization. This deficiency appears to be a feature of all currently existing exchange-correlation functionals, the key approximation in DFT. As QMC is a more accurate electronic structure method, barring discovery of an error in our implementation and execution, we expect that our results will provide an invaluable data point for physicists, chemists, and materials scientists interested in developing better functionals.
The expectation that our QMC calculations are worthy of this benchmark status is born of the nature of these calculations, which involve the direct statistical sampling of high dimensional, many-body quantum mechanical quantities. These calculations are computationally very intensive; this entire study consumed approximately 20 million CPU hours split between Mira at ANL’s LCF and Sequoia at LLNL. This work was in collaboration with David Tománek’s group at Michigan State University.
[1] L. Shulenburger, et. al., “The Nature of the Interlayer Interaction in Bulk and Few-Layer Phosphorus”, Nano Letters, 2015.
[2] L. Cartz, et. al., “Effect of pressure on bonding in black phosphorus”, The Journal of Chemical Physics, 1983.
[3] H. Liu, et. al., “Phosphorene: An Unexplored 2D Semiconductor with a High Hole Mobility”, ACS Nano, 2014.