Characterization of Materials and Chemical Processes
Introduction
The Sandia National Laboratories NMR Spectroscopy facility maintains both high resolution solution and solid state NMR capabilities for the measurement of chemical structure, reaction kinetics, morphologies and dynamic properties for a wide range of compounds and materials. Current efforts studies on polymers, composites, ceramics, glasses, lignins to CWA simulants. Efforts include the development and implementation of both multi-frequency and multi-dimensional NMR experiments designed to probe specific chemical and materials science related questions.
Structure and Dynamics in Polymers and Composites
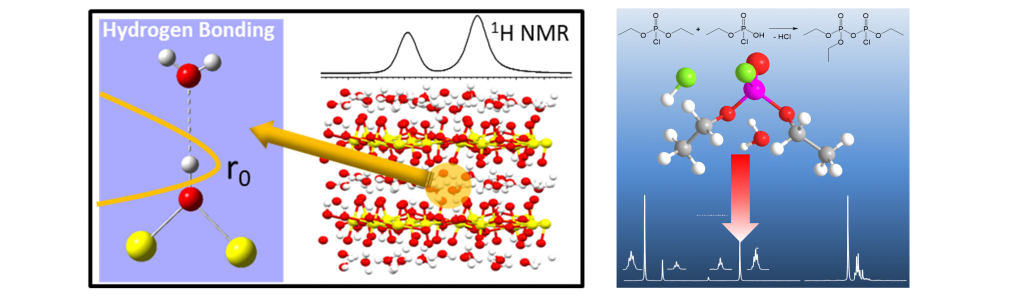
Understanding the local structure, morphology and dynamics in polymers and composites is crucial for the development and design of future materials tailored to address specific applications and performance designs. One of our current efforts involves the measurement of domain size and morphology in Sandia’s SDAPP (sulfonated Diels Alder Poly(Phenylene) proton exchange membranes (PEMs) and well as determining the Li coordination environment is ion-containing polyethylene polymers. We are especially focused on directly testing or providing input to computational simulations and model development for these same materials. Recent advances in Sandia’s NMR laboratory include the development of double quantum (DQ)-filtered and 1H-19F REDOR-filtered NMR spin diffusion experiments to address nano-scale morphology and local dynamics. We have also combined NMR chemical shift analysis with DFT cluster calculations to directly measure hydrogen bond strength in various polymer systems.
- Brad H. Jones, Todd M. Alam, Sangwoo Lee, Mathew C. Celina, Joshua P. Allers, Sungmin Park, Liwen Chen, Estevan J. Martinez, Jaclynn L. Unangst, ”Curing behavior, chain dynamics, and microstructure of high Tg thiol-Acrylate networks with systematically varied network heterogeneity”, Polymer, 122783, (2020), https://doi.org/10.1016/j.polymer.2020.122783.
- Todd M. Alam, Joshua P. Allers, Brad H. Jones, “Heterogeneous Polymer Dynamics Explored Using Static 1H NMR Spectra”, Int. J. Mol. Sci., 21, 5176 (2020), https://doi.org/10.3390/ijms21155176
- Todd M. Alam and Brad H. Jones, “Investigating Chain Dynamics in Highly Crosslinked Polymers using Solid‐State 1H NMR Spectroscopy”, Journal of Polymer Science, Part B Polymer Physics, Early View (2019). https://onlinelibrary.wiley.com/doi/abs/10.1002/polb.24869
- Todd M. Alam, “Chapter 4: High Resolution Magic Angle Spinning (HRMAS) Pulse Field Gradient (PFG) NMR Diffusometry Studies of Swollen Polymers”, in NMR Methods for Characterization of Synthetic and Natural Polymers”, Ed. R. Zhang, T. Miyoshi and P. Sun, pg. 63-79 (2019). https://pubs.rsc.org/en/content/ebook/978-1-78801-400-7
- Eric G. Sorte, Benjamin A. Paren, Christina G. Rodriguez, Cy Fujimoto, Cassandria Poirier, Lauren J. Abbott, Nathaniel A. Lynd, Karen I. Winey, Amalie L. Frischknecht, and Todd M. Alam, “Impact of Hydration and Sulfonation on the Morphology and Ionic Conductivity of Sulfonated Poly(phenylene) Proton Exchange Membranes”, Macromolecules 52, 857-876 (2019). DOI: 10.1021/acs.macromol.8b02013
- Eric G. Sorte, Lauren J. Abbott, Amalie L. Frischknecht, Mark Wilson, and Todd M. Alam, “Hydrophilic Domain Structure in Polymer Exchange Membranes: Simulations of NMR Spin Diffusion Experiments to Address Ability for Model Discrimination”, J. Polymer Science Part B: Polymer Physics, 56, 62-78 (2018). DOI:10.1002/polb.24439
- Todd M. Alam, “Computational Study of Microhydration in Sulfonated Diels–Alder Poly(phenylene) Polymers”, J. Phys. Chem. A, 122, 3927-3938 (2018) DOI: 10.1021/acs.jpca.8b0135
- Eric G. Sorte and Todd M. Alam, “1H-19F REDOR-Filtered NMR Spin Diffusion Measurements of Domain Size in Heterogeneous Polymers” Magn. Reson. Chem., 1–9 (2017). https://doi.org/10.1002/mrc.4623
- Todd M. Alam and Michael R. Hibbs, “Characterization of Heterogeneous Solvent Diffusion Environments in Anion Exchange Membranes”, Macromolecules, 47, 1073-1084 (2014). http://dx.doi.org/10.1021/ma402528v
- C. Francisco Buitrago, Janelle E. Jenkins, Kathleen L. Opper, Brian S. Aitken, Kenneth B. Wagener, Todd M. Alam, and Karen I. Winey, “Room Temperature Morphologies of Precise Acid- and Ion-Containing Polyethylenes”, Macromolecules, 46(22), 9003-9012 (2013). http:/dx/doi.org/10.1021/ma4013169
- C. Francisco Buitrago, Todd M. Alam, Kathleen L. Opper, Brian S. Aitken, Kenneth B. Wagener, and Karen I. Winey, “Morphological Trends in Precise Acid- and Ion-Containing Polyethylenes at Elevated Temperature”, Macromolecules, 46(22),8995-9002 (2013). http:/dx/doi.org/10.1021/ma4013175
- Todd M. Alam, Janelle E. Jenkins, Dan S. Bolinitineanu, Mark J. Stevens, Amalie L. Frischknecht, C. Francisco Buitrago, Karen I. Winey, Kathleen L. Opper and Kenneth B. Wagener, “Heterogeneous Coordination Environments in Lithium-Neutralized Ionomers Identified Using 1H and 7Li MAS NMR”, Materials, 5, 1508-1527 (2012). http://www.mdpi.com/1996-1944/5/8/1508
NMR Diffusometry
NMR provides the perfect tool to transport properties of solvents and abdsorbed chemicals in materials. Through the use of NMR diffusometry studies using pulse field gradient (PFG) NMR experiments it is possible to directly measure diffusion of individual specie sin complex mixtures. The laboratory has also been developing the use of high resolution magic angle spinning (HRMAS) PFG NMR to look at diffusion in heterogeneous materials. This evolving technique can find a wide applicaiton of uses in material, chemical and physical studies.
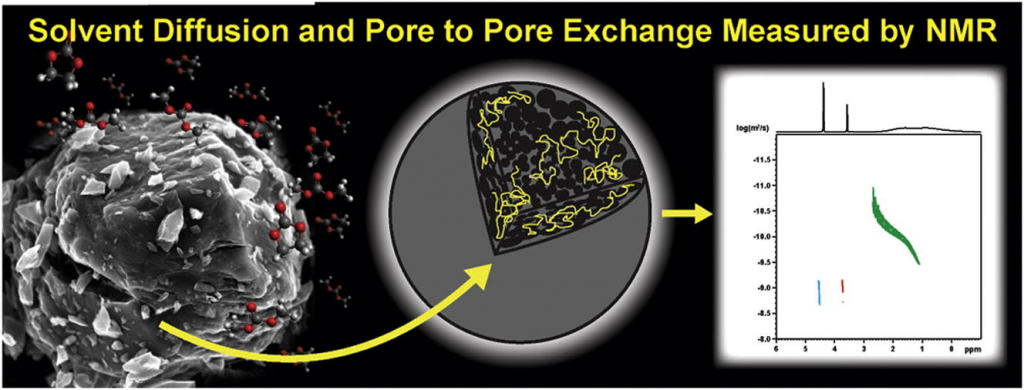
- Todd M. Alam, “Chapter 4: High Resolution Magic Angle Spinning (HRMAS) Pulse Field Gradient (PFG) NMR Diffusometry Studies of Swollen Polymers”, in NMR Methods for Characterization of Synthetic and Natural Polymers”, Ed. R. Zhang, T. Miyoshi and P. Sun, pg. 63-79 (2019). https://pubs.rsc.org/en/content/ebook/978-1-78801-400-7 or click image below.

- Todd M. Alam and Michael R. Hibbs, “Characterization of Heterogeneous Solvent Diffusion Environments in Anion Exchange Membranes”, Macromolecules, 47, 1073-1084 (2014). http://dx.doi.org/10.1021/ma402528v
- Todd M. Alam and Janelle E. Jenkins, “ HR-MAS NMR Spectroscopy in Material Science”, in Advanced Aspects of Spectroscopy, Muhammad Akhyar Farrukh (Ed.), ISBN: 978-953-51-0715-6, InTech, (2012) Chapter Available:
- Todd M. Alam and Thomas M. Osborn Popp, “In-Pore Exchange and Diffusion of Carbonate Solvent Mixture in Nanoporous Carbon” Chem. Phys. Lett., 658, 51-57 (2016). doi:10.1016/j.cplett.2016.06.014
- Janelle J. Jenkins, Michael R. Hibbs and Todd M. Alam, “Identification of Multiple Diffusion Rates in Mixed Solvent Anion Exchange Membranes Using High Resolution MAS NMR”, ACS Macro Letters, 1, 910-914 (2012).paper
- Todd M. Alam, Kimberly K. Childress, Kevin Pastoor, Charles V. Rice, “Characterization of Free, Restricted and Entrapped Water Environments in Poly(N-Isopropyl Acrylamide) Hydrogels via 1H HRMAS PFG NMR Spectroscopy”, J. Polymer Science: Polymer Physics, 52 1521-1527 (2014) http://dx.doi.org/10.1002/polb.23591
- Todd M. Alam, Daniel R. Dreyer, Christopher W. Bielawski, and Rodney S. Ruoff, “Combined Measurement of Translational and Rotational Diffusion in Quantenary Acyclic Ammonium and Cyclic Pyrrolidinium Ionic Liquids”, J. Phys. Chem. B, 117, 1967-1977 (2013). http://dx.doi.org/10.1021/jp3111953